
Vascularite urticarienne hypocomplémentémique
La vascularite urticarienne est une entité anatomo-clinique définie par une atteinte inflammatoire des capillaires du derme associée à une éruption cutanée urticarienne atypique.
INTRODUCTION
La vascularite urticarienne (VU) est une entité anatomo-clinique définie par une atteinte inflammatoire des capillaires du derme associée à une éruption cutanée urticarienne atypique. L’atteinte cutanée, comme elle est classiquement décrite, est faite d’éléments non fugaces, violacés, associés à une sensation de prurit, de douleur ou de brûlure durant plus de 24 heures et laissant volontiers une pigmentation résiduelle. Elle correspond sur le plan histologique à une vascularite leucocytoclasique, caractérisée par une atteinte vasculaire avec un infiltrat périvasculaire fait majoritairement de polynucléaires neutrophiles (PNN), le plus souvent à noyaux pycnotiques définissant la leucocytoclasie.
Les VU représentent un spectre très étendu allant de mani- festations cutanées isolées à des manifestations systémiques potentiellement sévères, avec de nombreuses formes intermé- diaires. Les VU peuvent être divisées en 2 groupes selon le taux des fractions du complément : vascularites urticariennes normocomplémentémiques (VUN) et vascularites urticariennes hypocomplémentémiques (VUH). Alors que la grande majorité des VUN semblent idiopathiques, des cas de maladies systémiques comme le lupus érythémateux systémique (LES), le syndrome de Sjögren et certaines hémopathies ont été rapportés dans le cadre de VUH. En l’absence de maladie sous-jacente identifiée, la VUH était classiquement considérée dans la littérature comme idio- pathique et semblait représenter un désordre immunitaire rare, systémique, appelé syndrome de vascularite urticarienne hypo- complémentémique (SVUH) ou vascularite de McDuffie.
Ainsi, la nomenclature et la classification des VU ont fait l’objet de nombreux débats depuis des décennies. À l’heure actuelle, la relation entre VUN, VUH et SVUH n’est pas clairement définie. Bien que non univoque, il semble exister un continuum entre la VUH et le SVUH. La nomenclature de Chapel Hill, révisée en 2012, indivi- dualise la VUH sous le terme de vascularite avec anticorps anti-C1q, définie par une vascularite touchant les vaisseaux de petit calibre, accompagnée de lésions urticariennes et d’une hypocomplémen- témie [4]. Les atteintes articulaires, pulmonaires et oculaires sont fréquentes. La présence d’anticorps anti-C1q est caractéristique bien qu’inconstante et non pathognomonique. En accord avec cette nomenclature, nous parlerons dans ce travail de VUH au sens large, qu’elles soient associées ou non à une maladie sous-jacente.
EPIDEMIOLOGIE
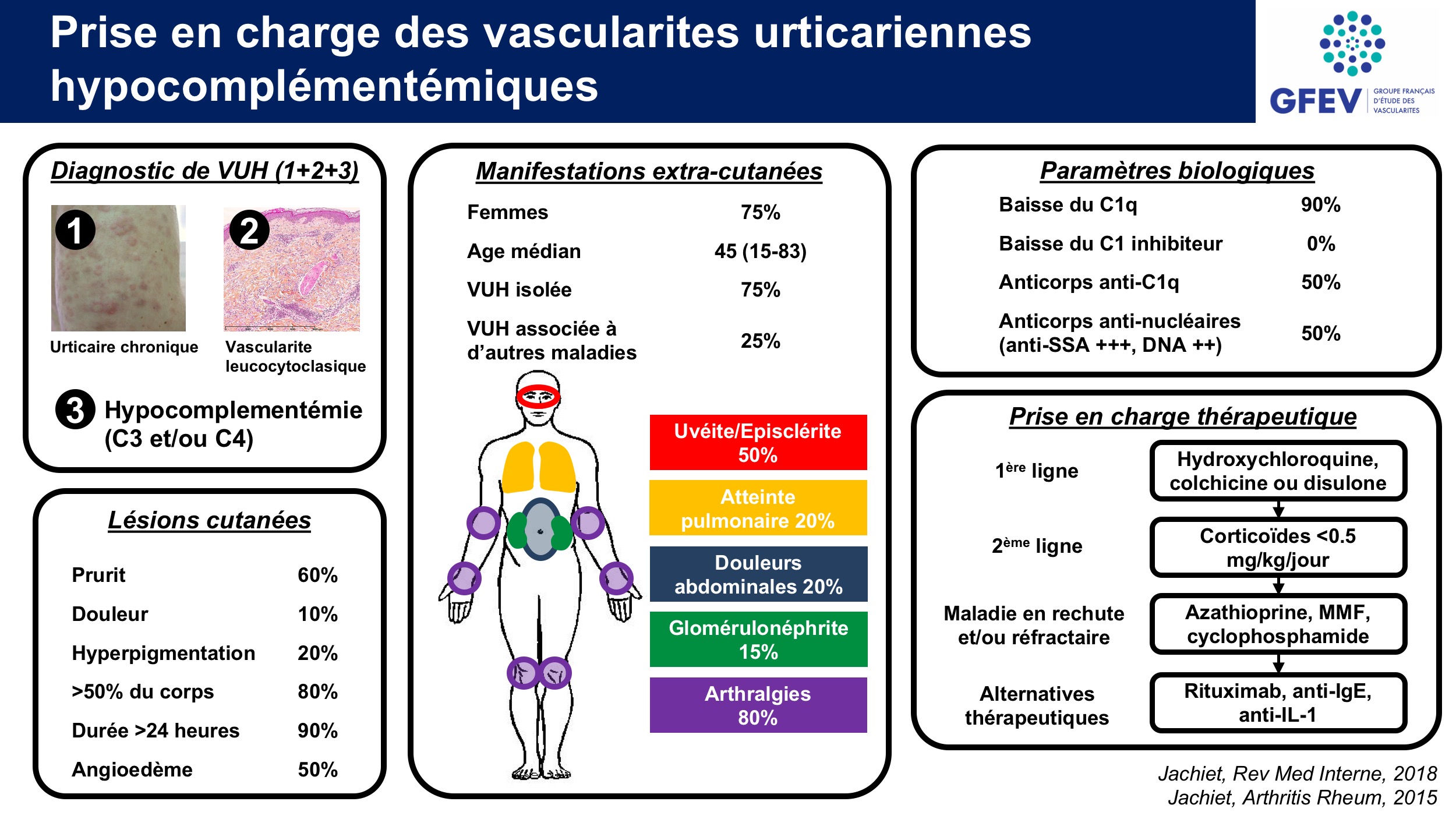
La VUH est une entité rare. La prévalence et l’incidence sont inconnues et difficiles à évaluer, du fait de la diversité des manifes- tations cliniques, des difficultés de diagnostic et de classification et du faible nombre de patients. La revue de la littérature entre 1970 et 2016 fait état de près de 260 cas rapportés. Nous avons récem- ment rapporté une série rétrospective nationale de 57 patients ayant une VUH diagnostiquée entre 1992 et 2014 dans 27 centres francais. Le délai entre les premières manifestations cliniques et le diagnostic varie de 18 à 68 mois. Quelques séries rapportent une prévalence de 2,7 à 50 % de VUH au sein du groupe des VU. L’âge moyen au diagnostic était de 47 ans (extrêmes 15–83) dans notre série. Il existait une nette prédomi- nance féminine à 74 %. Plus rares, quelques cas pédiatriques sont rapportés, avec la particularité de présenter une atteinte rénale plus fréquente.
MALADIES ASSOCIEES
La VUH est isolée dans 75 %, et peut être associée à une maladie sous-jacente dans 25 % des cas. Dans ce dernier cas, il s’agit prin- cipalement de maladies systémiques, au premier rang desquelles se situe le LES dans l’immense majorité des cas, dans la littérature comme dans notre série. De façon anecdotique, on trouve quelques cas de syndrome de Sjögren, de sclérodermie systémique, de polyarthrite rhumatoïde, de cryoglobulinémie associée à une hépatite C, de polychondrite atrophiante, ou de maladie associée aux IgG4. De très rares cas d’hémopathie lymphoïde ou de cancer solide sont décrits. Dans la série récente que nous avons publiée comme dans la littérature, le très petit nombre de patients atteints de cancer et/ou d’hémopathie maligne ne permet pas de retenir une association entre le cancer et les VUH . Ces données soulignent l’importance de rechercher un lupus asso- cié devant une VUH, mais il n’est pas justifié de réaliser un dépistage systématique de cancer ou d’hémopathie maligne.
DIAGNOSTIC
La description princeps faite par McDuffie et al. en 1973 rapportait les cas de 4 patients ayant une vascularite cutanée avec arthrites et hypocomplémentémie (baisse du C3, C4, CH50 et C1q pour les 4 patients). En 1982, Schwartz et al. ont proposé des critères diagnostiques. La VUH était définie par 2 critères majeurs : lésions urticariennes chroniques et hypocomplémentémie, et au moins 2 des 6 critères mineurs : vascularite leucocytoclasique, arthralgies ou arthrites, inflammation oculaire, glomérulonéphrite, douleurs abdominales et anticorps anti-C1q.
PHYSIOPATHOLOGIE
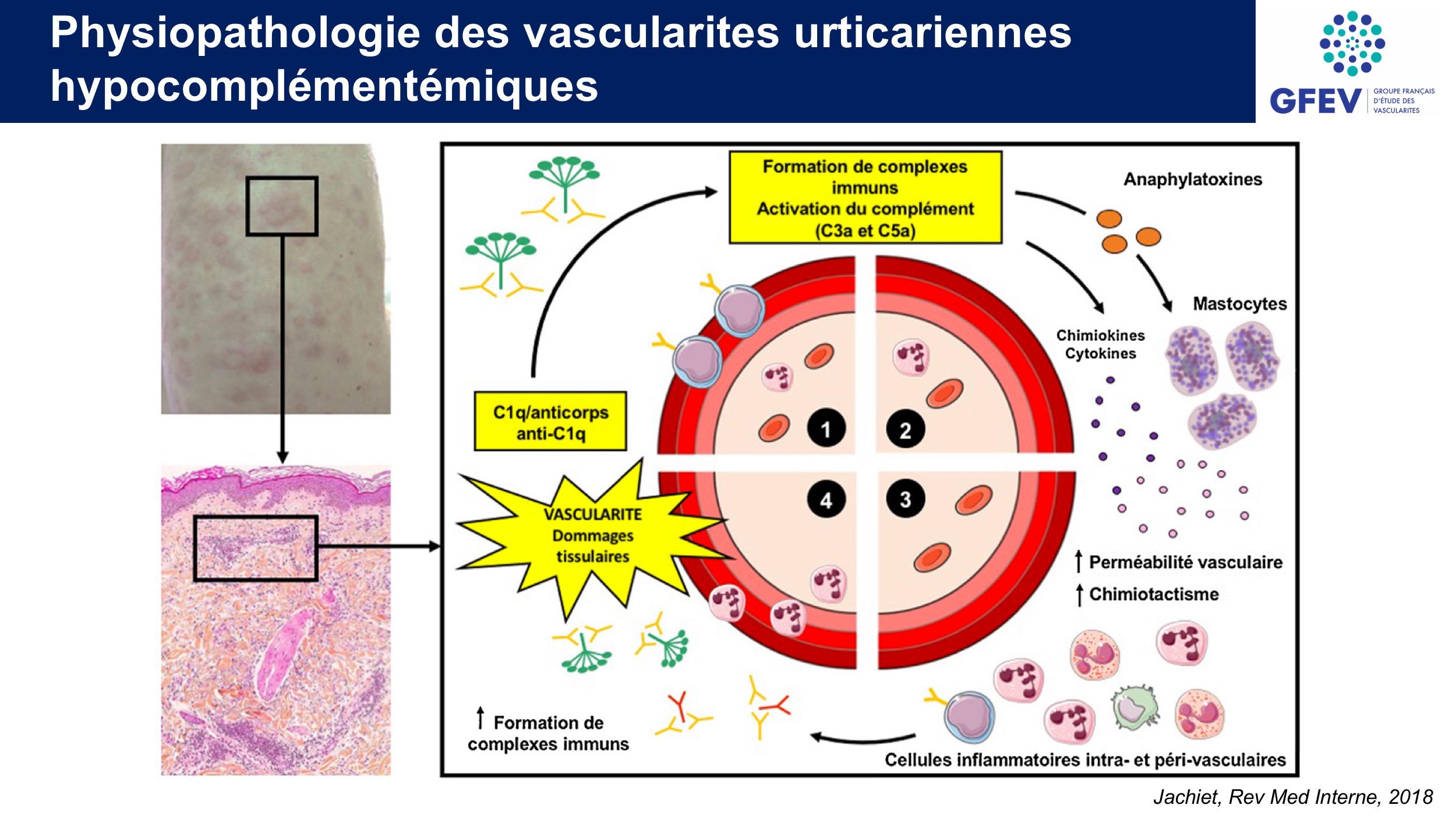
La VUH est une vascularite médiée par les complexes immuns et correspond à une réaction d’hypersensibilité de type III. Les mécanismes possibles des dommages vasculaires impliqueraient les complexes immuns, les anticorps anti-C1q ainsi qu’une réponse médiée par les lymphocytes T.
Les complexes immuns C1q-anticorps anti-C1q, présents dans le sérum, activeraient la voie classique du complément au niveau des vaisseaux. L’activation du complément génèrerait la libération de cytokines, chimiokines, anaphylatoxines et la dégranulation des mastocytes. Ces substances majoreraient alors la perméabilité vas- culaire, le chimiotactisme des cellules inflammatoires et le dépôt des complexes immuns, favorisant l’œdème et la destruction des parois vasculaires.
Les anticorps anti-C1q ne sont pas spécifiques des VUH et ont été décrits dans d’autres maladies comme le LES (où ils sont corrélés à l’hypocomplémentémie et l’atteinte rénale), le syndrome de Sjögren, la polyarthrite rhumatoïde, le syndrome de Goodpasture, la sclérodermie, la cryoglobulinémie, la néphropathie à IgA, la glomérulonéphrite aiguë post-streptococcique ou d’autres connectivites.
ATTEINTE CUTANEE
L’atteinte cutanée, constante, est faite de papules érythémateuses et œdémateuses « urticariennes », de topographie diffuse sur le tronc et les membres. Dans la grande majorité des cas, elles sont fixes, durent plus de 24 heures et laissent une pigmenta- tion résiduelle post-inflammatoire. Classiquement décrites comme douloureuses, les lésions étaient prurigineuses dans 60 % dans cas et douloureuses dans 11 % des cas dans notre série. La fréquence des angioœdèmes est proche de 50 %. Ils sont localisés principalement sur le visage, les paupières, les lèvres, la langue, rarement le larynx. Une atteinte menaçante a été exceptionnellement rapportée dans la littérature. Du purpura était présent dans 35 % et un livedo dans 14 % des cas dans notre série. Une photosensibilité est décrite dans 14 % des cas.
L’image histologique classique est celle d’une vascularite leucocytoclasique. Sous un épiderme normal, le derme, parfois œdémateux (30 %), est le siège d’un infiltrat inflammatoire polymorphe. L’infiltrat, périvasculaire (80 %) et/ou interstitiel (50 %), est principalement composé de PNN (100 %), avec leucocytoclasie dans 95 % des cas, mais également de lymphocytes, de polynucléaires éosinophiles (PNE) et d’histiocytes. Dans notre série, les lésions histologiques étaient généralement moins franches que dans les quelques séries antérieures détaillant l’histologie. Les altérations vasculaires étaient moins marquées, avec une fréquence moindre de nécrose fibrinoïde (25 %), d’œdème des cellules endothéliales et d’extravasation d’hématies. Il semble important de répéter les biopsies, car les signes histologiques peuvent varier dans le temps, sur le type et sur l’intensité des lésions. L’immunofluorescence directe (IFD) est positive dans plus de la moitié des cas, avec des dépôts péri-capillaires (87 %) et/ou sur la jonction dermo-épidermique (67 %), constitués principalement de C3 et IgG, plus souvent que d’IgM, IgA et C1q. Des dépôts granuleux d’IgG à la jonction dermo-épidermique évocateurs de bande lupique sont décrits, tant pour les VUH isolées que pour les VUH associées au LES.
ATTEINTES EXTRA-CUTANEES
En plus de l’atteinte cutanée, 68 % des patients de notre série avaient une VUH systémique, définie par la présence d’au moins deux atteintes d’organes parmi les atteintes oculaire, articulaire, digestive et/ou rénale. Le nombre médian d’organes atteints en dehors de la peau était de 2 (0–4), alors qu’il est rarement précisé dans la littérature.
Les signes généraux étaient présents dans plus de 50 % des cas dans notre série, à type d’asthénie et de fièvre lors des poussées.
L’atteinte articulaire était présente dans plus de 80 % des cas dans notre série. Elle se caractérise par des arthralgies, générale- ment transitoires et touchant principalement les coudes, poignets, genoux et chevilles, parfois des arthrites ou des synovites, rarement des myalgies. Deux cas de myosite ont été décrits. L’atteinte articulaire est non déformante, à l’exception de l’arthropathie de Jaccoud, rapportée dans moins de 10 cas. Dans le cadre des VUH, l’arthropathie de Jaccoud s’associe constamment à une atteinte valvulaire cardiaque ou vasculaire.
L’atteinte pulmonaire, présente dans 19 % des cas dans notre série, est inférieure aux séries antérieures, proche de 50 %. Cette différence pourrait en partie être liée à un tabagisme actif ou passif plus fréquent, qui concernaient 91 à 100 % des patients ayant une atteinte pulmonaire dans deux séries anté- rieures comparativement à 36 % dans notre série. La dyspnée et la toux sont les symptômes principaux. Les douleurs thoraciques, les expectorations, l’hémoptysie, l’hémorragie intra-alvéolaire et l’épanchement pleural ont été rarement rapportés. Les épreuves fonctionnelles respiratoires (EFR) révèlent un trouble ventilatoire obstructif (TVO) dans près de 50 % des cas qui peut être initialement asymptomatique. Il évolue lentement et conduit rarement à la transplantation pulmonaire ou au décès. Des atteintes obstructives sévères ont été rapportées de fac ̧on précoce même en l’absence consommation tabagique, mais la majorité des patients ayant une atteinte pulmonaire spé- cifique de VUH fume. De plus, la consommation de tabac semble corrélée à la sévérité des lésions. Un emphysème pulmonaire est présent dans moins de 50 % des cas. Bien souvent asymptomatique initialement, il survient de fac ̧ on assez précoce (7,2 ans en moyenne après le début des lésions urticariennes dans la série de Schwartz et al.). L’atteinte pulmonaire obstruc- tive pourrait résulter de l’association d’une vascularite pulmonaire (inflammation, élastinolyse, destruction protéolytique du parenchyme) et d’un emphysème, avec un rôle potentiel aggravant du tabac. Les anticorps anti-C1q pourraient contribuer à l’atteinte obstructive en plus de la vascularite des capillaires et vei- nules pulmonaires, en se liant à des protéines du surfactant des alvéoles régulant la tension à la surface des alvéoles et entraînant la destruction des alvéoles.
L’atteinte rénale était présente dans 14 % des cas dans notre série. Le plus souvent modérée, quelques cas d’insuffisance rénale terminale menant à la dialyse voire à la transplantation rénale sont rapportés dans la littérature. Elle semble plus fréquente et plus sévère chez l’enfant. L’atteinte rénale est principalement glomérulaire, révélée par une protéinurie modérée et une hématurie, mais pouvant aller jusqu’au syndrome néphrotique et à l’insuffisance rénale constituée.
L’atteinte digestive se manifeste principalement par des douleurs abdominales, diarrhée, nausées et vomissements. Quelques cas d’ascite ont été rapportés, et de façon anecdotique, un cas de pancréatite aiguë et une localisation de vascularite digestive dans la vésicule biliaire.
Une atteinte oculaire inflammatoire spécifique est classique au cours des VUH, présente dans 56 % des cas de notre série. Elle se manifeste par ordre de fréquence par une conjonctivite, une sclérite ou épisclérite, ou une uvéite. Généralement transitoire et réversible, le pronostic visuel est bon, en dehors de très rares cas d’atrophie du nerf optique ou de cécité.
D’autres atteintes d’organes sont possibles au cours de cette vascularite systémique.
CARACTERISTIQUES BIOLOGIQUES
L’immense majorité des patients a une hypocomplémentémie, avec baisse du C3, C4 et/ou CH50. Seulement la moitié des patients de notre série avaient des anticorps anti-C1q détectables lorsqu’ils étaient recherchés. Plusieurs séries précédentes montraient une fréquence allant de 0 % à 100 % selon les critères d’inclusion choisis par les auteurs. Leur absence pourrait en partie être expliquée par des difficultés techniques, une clairance rapide des anticorps ou leur dépôt dans les tissus. En revanche, la très grande majorité des patients (90 à 100 %) avait une diminution du taux de C1q, ce qui suggère qu’un taux de C1q abaissé ou effondré dans le cadre d’une VU serait un marqueur plus sensible que les anticorps anti-C1q pour le diagnostic de VUH. L’inhibiteur de la C1 estérase est constamment normal. La majorité des patients présentait un profil d’activation de la voie classique du complément, impliquant probablement des complexes immuns circulants C1q-anticorps anti-C1q. Par ailleurs, les anticorps anti-C1q semblaient être associés à un phénotype clinique particulier, avec un angioœdème, un livedo, des atteintes oculaire, articulaire et rénale plus fréquentes et des atteintes pulmonaire et digestive moins fréquentes. En revanche, la forme systémique de VUH n’était pas corrélée à un taux inférieur de complément par rapport à la forme non systémique.
Les anticorps anti-nucléaires (AAN) étaient positifs dans 50 % des cas de notre série, en accord avec les données antérieures.
TRAITEMENT
La prise en charge thérapeutique est mal codifiée et souvent peu détaillée dans la littérature. De multiples traitements ont été essayés de fac ̧ on empirique, selon les données de cas cliniques et de séries, en l’absence de traitement spécifique et de recommandations thérapeutiques précises.
Dans notre série, tous les patients ont reçu au moins une ligne de traitement antihistaminique, constamment inefficace sur les lésions cutanées et les manifestations extra-cutanées, malgré l’augmentation de la posologie et les changements ou associations de molécules. En dehors des antihistaminiques, le nombre médian de traitements reçus était de 3 (extrêmes 1–9), et 50 % des patients ont reçu plus de 3 lignes de traitement. La durée médiane de rémis- sion était de 6 à 11 mois (extrêmes 2–72 mois) en fonction de la ligne de traitement.
En première ligne thérapeutique, les patients ont reçu principalement des corticoïdes (57 %) à une dose médiane initiale de prednisone de 40 mg/j (extrêmes 10–80 mg/j), de l’hydroxychloroquine (HCQ) (46 %) et de la colchicine (14 %). Les taux de réponse étaient comparables avec l’HCQ (50 % de réponse cutanée et 48 % de réponse immunologique), la colchicine (43 % de réponse cutanée et 40 % de réponse immunologique), la dapsone (100 % de réponse cutanée et 0 % de réponse immunologique) et les corticoïdes (53 % de réponse cutanée et 32 % de réponse immunologique). Ainsi, en première intention, l’HCQ, la colchicine et la dapsone semblent représenter une option thérapeutique valable, avec une efficacité similaire à celle des corticoïdes et des effets secondaires moindres. L’efficacité des corticoïdes était similaire, qu’ils aient été utilisés à une dose supérieure ou inférieure à 20 mg/jour.
Les patients ayant une maladie réfractaire et/ou en rechute ont reçu principalement des corticoïdes et des immunosuppresseurs, notamment l’azathioprine (AZA), le méthotrexate (MTX), le mycophénolate mofétil (MMF) ou le rituximab (RTX). Chez les patients ayant une maladie réfractaire et/ou en rechute, les taux de réponse étaient légèrement supérieurs lorsque les corticoïdes étaient associés aux immunosuppresseurs conventionnels, en particulier l’AZA (80 % de réponse cutanée et 73 % de réponse immunologique), le MMF (56 % de réponse cutanée et 40 % de réponse immunologique), le MTX (67 % de réponse cutanée et 50 % de réponse immunolo- gique) ou le CYC (86 % de réponse cutanée et 100 % de réponse immunologique) versus les corticoïdes (65 % de réponse cutanée et 46 % de réponse immunologique). Toutefois, un effet d’épargne cortisonique est probable lorsque les immunosuppresseurs sont associés à la corticothérapie.
Quelle que soit la ligne thérapeutique, il est observé une association significative entre la réponse cutanée et la réponse immunologique correspondant aux taux de complément. Cela souligne l’intérêt de suivre l’évolution des taux de complément au cours du traitement, car il pourrait s’agir d’un marqueur prédictif de la réponse thérapeutique, mais également d’un marqueur précé- dant la survenue d’une rechute clinique. La réponse extra-cutanée était parallèle à la réponse cutanée dans 80 % des cas. Les taux de réponse extra-cutanée les plus faibles concernaient principalement l’atteinte oculaire à type de sclérite et d’uvéite et l’atteinte pulmonaire, qui représentaient les manifestations cliniques les plus réfractaires. Enfin, contrairement à la présentation clinique, la présence ou l’absence d’anticorps anti-C1q n’avait aucune incidence sur les taux de réponse ni sur la durée médiane avant échec du traitement.
CONCLUSION
En conclusion, la VUH est une vascularite systémique rare avec diverses manifestations cliniques, caractérisées principalement par des atteintes articulaire, oculaire et des angioœdèmes dans plus de la moitié des cas, en plus de l’atteinte cutanée qui est constante. Plus de deux tiers des patients ont une VUH systémique avec au moins deux atteintes d’organes parmi les atteintes oculaire, arti- culaire, digestive et/ou rénale. Les anticorps anti-C1q sont détectés seulement chez la moitié des patients, aussi le taux abaissé ou effon- dré de C1q semble représenter un marqueur plus sensible pour le diagnostic de VUH. Toutefois, la présence des anticorps anti-C1q semble identifier un phénotype de patients particulier, avec des atteintes oculaire, articulaire, rénale, un angioœdème et un livedo plus fréquents et des atteintes pulmonaire et digestive moins fréquentes.
Présentation sur les vascularites urticariennes

Fiches sur les vascularites urticariennes hypocomplémentémiques